[Seeding@Biophysics]$ Protein Dynamics and Functions
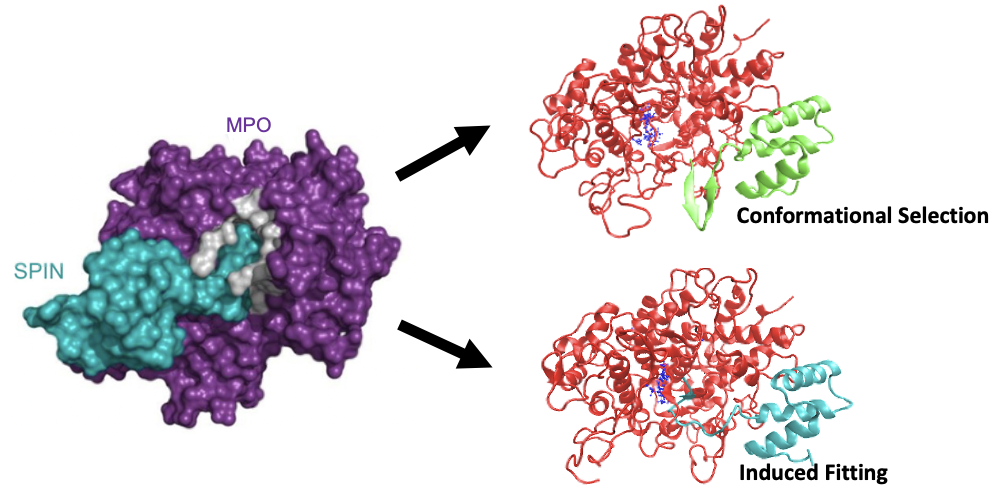
Staphylococcal Peroxidase INhibitor, SPIN, can bind to Human Myeloperoxidase (MPO) enzyme and inhibits the enzyme activities to prevent immune killing.
Studies have identified a disorder-to-order transition on SPIN's N-terminal domain (NTD), which functions to inhibit the enzyme activity. However, the details of SPIN's functional mechanisms lack understanding.
Here, we studied two SPIN homologs that present high sequence identity but different inhibitory abilities. By performing atomistic simulations, we discovered different binding and folding mechanisms adopted by two homologs, which could help to explain their functional discrepancies. Read more here.
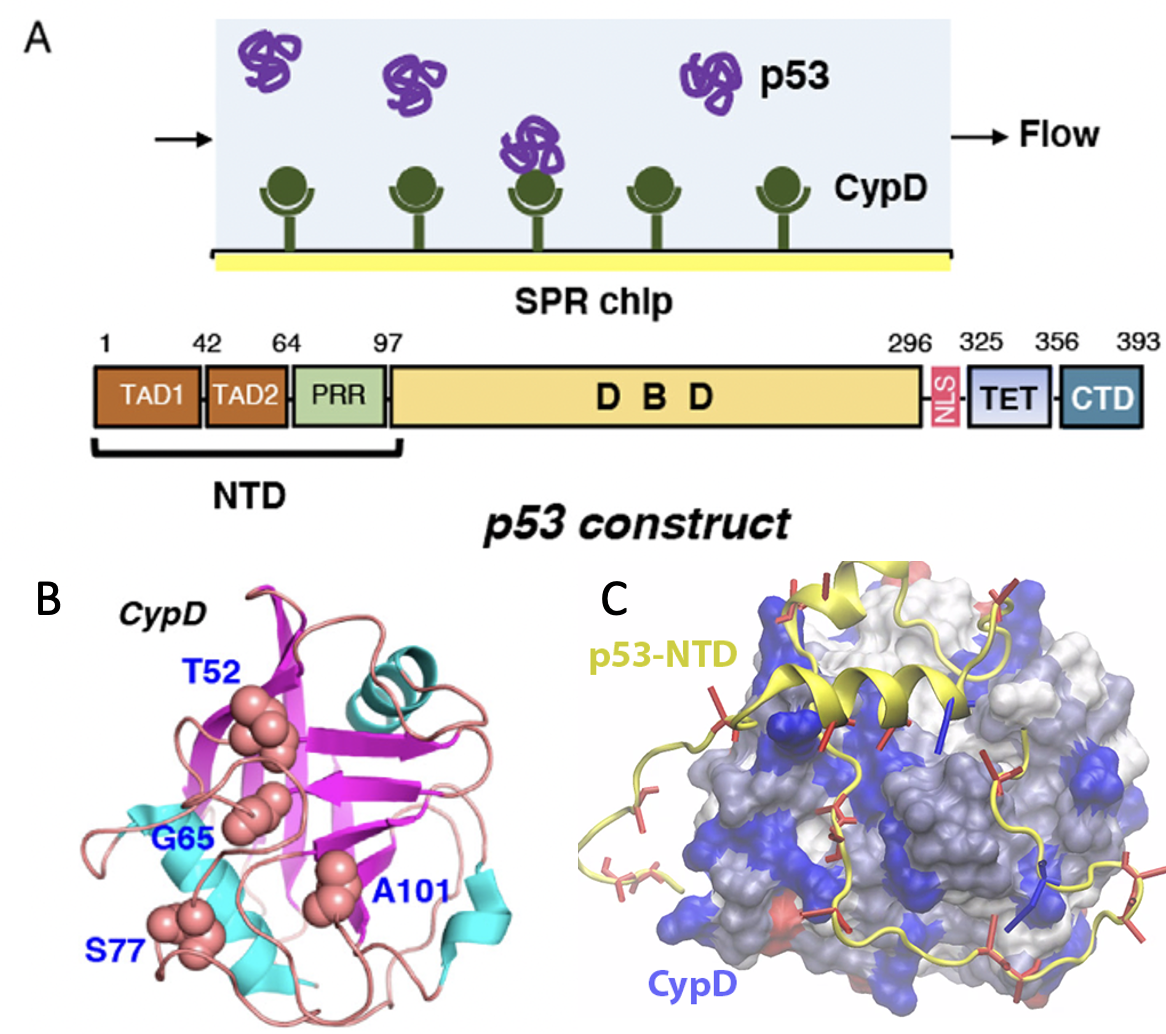
Cyclophilin D (CypD) is one crucial confirmed component of mitochondrial permeability transition pore (mPTP) regulator, which will be regulated by p53 N-terminal domain (p53-NTD).
Here, we studied the dynamic interactions between p53-NTD and CypD. We observed a p53-NTD binding surface on CypD, which were further identified by NMR experiments. The electornic interactions are found to be the driven forces for CypD/p53-NTD interactions. Read more here.
Liquid-liquid phase separation (LLPS) is one critical phenomena populated in cells that help biomolecules organization, regulation, and function. There are many signficant factors that may affect biomolecular phase separations. Particularly, the secondary structure dynamics play significant roles in regulating LLPS.
Here, we studied how the dynamics of IDP secondary structures could affect their LLPS properties.
Our simulations suggests how mutations could affect LLPS by affecting the protein secondary strcutures. Read more here.
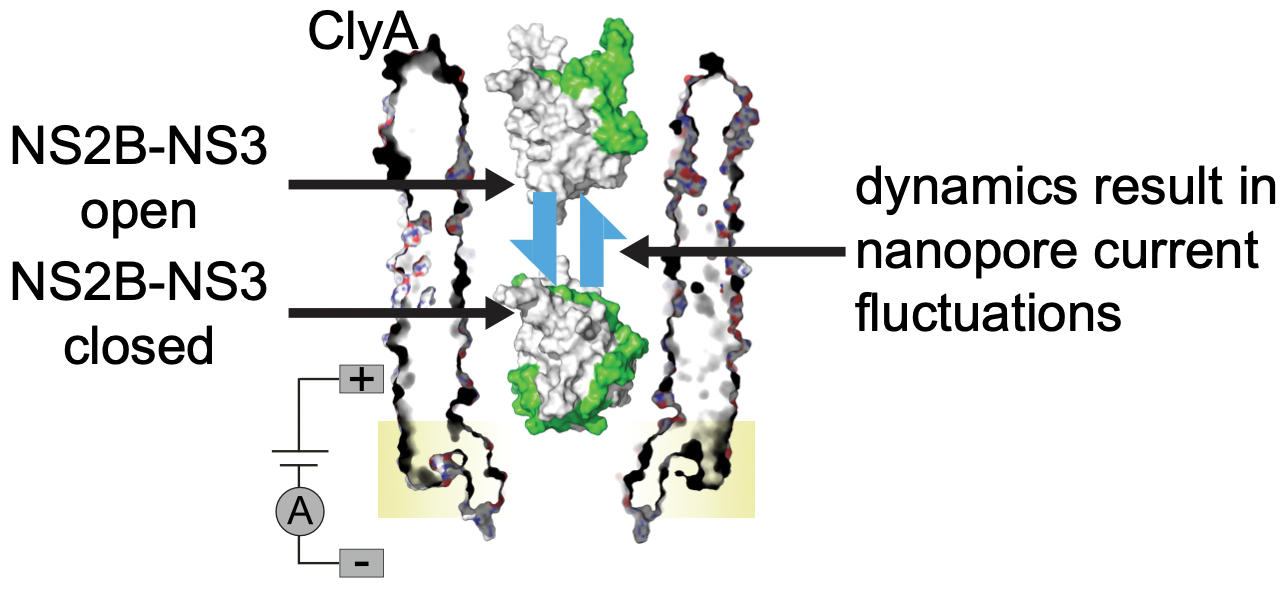
The nanopore tweezer is advanced in capturing the conformational dynamics of protein at single molecular level! MD simulations, on the other hand, can offer the significant molecular insights to resolve the current signal related conformations! The combination of two methods can achieve 1+1 >> 2 effects!
Here, we applied nanopore tweezer as a powerful tool to investigate the conformational dynamics of West Nile Virus NS2B/NS3 proteases, which are potential drug target for WNV infection treatments. Our simulations identified a significant protein binding position on ClyA, which could be specifically engineered to capture NS2B/NS3 dynamics regarding their functional activities. Read more here.
HyRes, the hydrid resolution coarse-grained model, was first developed by Dr. Liu, with qualitative predictions towards IDP secondary structures. The model has been further optimzied to more quantatatively to study IDP non-specific interactions and secondary structures.
HyRes II presents high accuracy and performance when studing long-time scale (like llps.) and complex system dynamics (like proteases in ClyA nanopore.).
If you are interested, the model can be achieved at:
HyRes II is now compiled with OpenMM and enabled GPU acceleration!
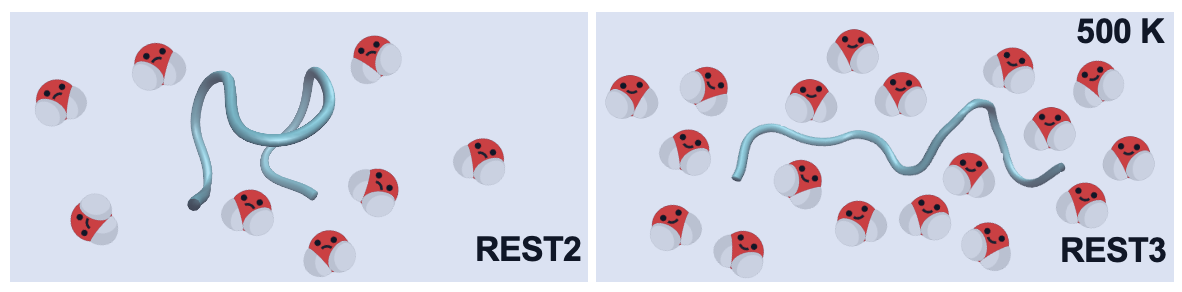
Replica Exchange with Solute Tempering (REST) is helpful in accelerating the explicited simulations. However, REST2 artifically scaled down the solute-solvent interactions under high temperature conditions, which over-compacted IDPs
Here, we treat the solue-solvent parameter freely and re-adjust it to approximatedly reproduce the optimal sampling under high temperatures. Read more here.
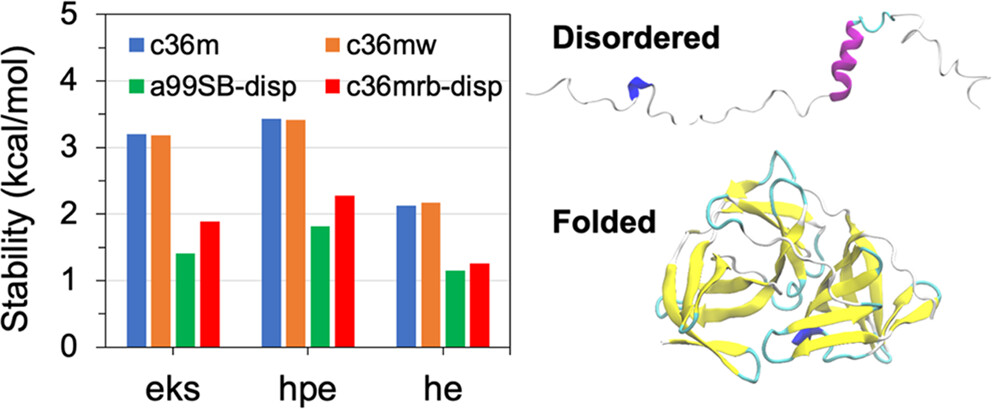
Recent years have witnessed drastic improvements in general-purpose explicit solvent protein force fields, partially driven by the need to study intrinsically disordered proteins (IDPs), and yet the state-of-the-art force fields such as CHARMM36m (c36m) and a99SB-disp still provide different performances in simulating disordered protein states, where c36m has a bias toward overcompaction for large IDPs.
Here, we examine the performance of c36m and a99SB-disp in describing the stabilities of a set of 46 amino acid backbone and side chain pairs in various configurations. We found that c36m systematically predicts stronger interactions compared to a99SB-disp. Guided by free energy decomposition, we evaluated if revising the charges alone could alleviate the severe overstabilization of salt bridges of c36m(w) vs a99SB-disp. The results suggested that the direct modification of protein–water interactions is also necessary. Toward this end, we proposed a tentative modification to c36m, referred to as c36mrb-disp, which combines modified Arg side chain charges, retuned backbone hydrogen bonding strength, and the a99SB-disp water model. Read more here.
[Seeding@ML&Modeling]$ Deep Learning Methodss
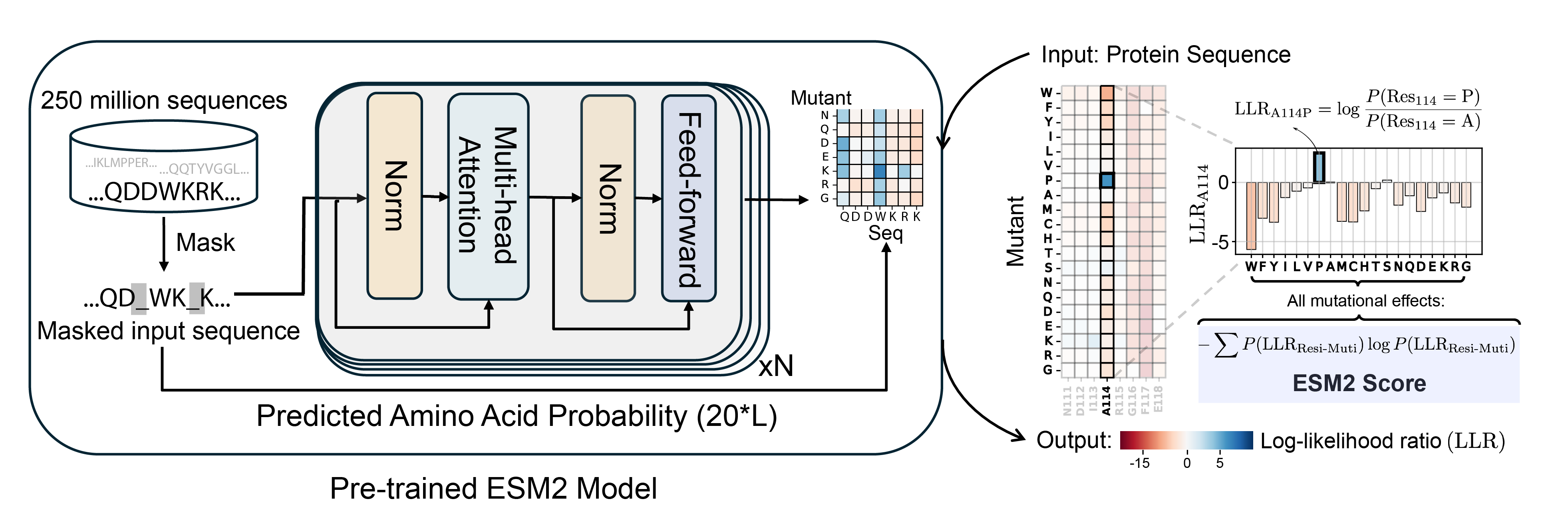
"Intrinsically disordered regions (IDRs) play a critical role in phase separation and are essential for the formation of membraneless organelles (MLOs). Mutations within IDRs can disrupt their multivalent interaction networks, altering phase behavior and contributing to various diseases. Therefore, examining the evolutionary fitness of IDRs provides valuable insights into the relationship between protein sequences and phase separation. In this study, we utilized the ESM2 protein language model to map the fitness landscape of IDRs. Our findings reveal that IDRs, particularly those actively participating in phase separation, contain conserved amino acids. This conservation is evident through mutational constraints predicted by ESM2 and supported by direct analyses of multiple sequence alignments. These conserved, disordered amino acids include residues traditionally identified as “stickers” as well as “spacers” and frequently form continuous sequence motifs. The strong conservation, combined with their critical role in phase separation, suggests that these motifs act as functional units under evolutionary selection to support stable MLO formation. Our findings underscore the insights into phase separation’s molecular grammar made possible through evolutionary analysis enabled by protein language models." Read more here.